Introduction
The congenital defect affecting the spectrum of oculo- auriculo- vertebral is characterised as the Goldenhar syndrome. This syndrome is associated with a constellation of overlapping anomalies that involve the face, eyes and ears.1 It was first described in the early 1950s and was also known as lateral facial dysplasia, Goldenhar complex or first and second branchial arch syndrome.2 Goldenhar syndrome (GS) goes unnoticed, since it shares its similarities with Hemifacialmicrosomia (HFM), hence it is also considered as a variant of HFM.2 GS was described by Maurice Goldenhar, but this rare entity was first recorded by Carl Ferdinand von Arlt.3 Goldenhar emphasized the syndrome as a triad of accessory tragus, mandibular hypoplasia, and limbaldermoids. Gorlin later named this syndrome as oculo‑auriculo‑vertebral dysplasia, due to the presence of additional vertebral anomalies. Due to the presence of vertebral defects in the syndrome as noted by Gorlin in 1963 it was also known as Goldenhar-Gorlin syndrome.1 The patho-physiologic hypothesis of this syndrome is explained in this article as the physical features vary based upon the severity of this syndrome.
Case Report
A two year old male child was referred to our department of Plastic Surgery, Saveetha Medical Hospital, Chennai diagnosed with secondary cleft palate. The parents complained that their child had frequent nasal regurgitation while consuming semi-solid to solid foods since feeding had been started. He is the only child born from a non-consanguineous marriage. No similar anomalies run in their family history. On general examination, the patient was active and co-operative. There was no history of alcohol or teratogenic drug intake by the mother. The child was born at full term by normal vaginal delivery. He cried immediately after birth and was breastfed for first 40 days of life and was gradually introduced to formula feeds. The child was evaluated by a diverse team of specialists including thepaediatric surgeon, paediatrician, ophthalmologist, otorhinolaryngologist, anesthesiologist, language and speech therapist, and occupational therapist. On general examination, the child weighed 14kgs with a normal blood picture and a haemoglobin of 10.2gm%. Facial asymmetry with unilateral hypoplastic mandible was evident, as well as frontal bossing over the right side of the face and minimal hair over the left eyebrow. There was a mild malar hypoplasia with hypoplastic facial muscles on the left side creating a decrease in function of facial expressions and mastFigure 1cation ($). All of the above mentioned facial features were reported by Robert. et al.4in 1963.$

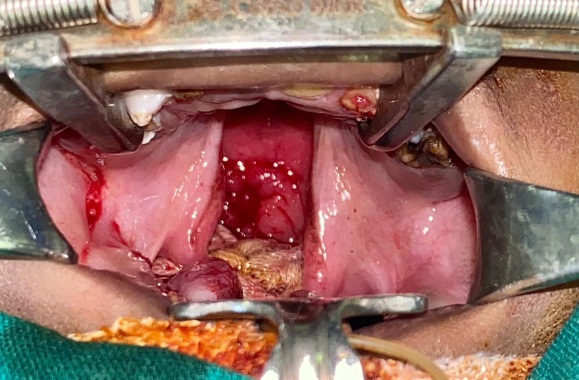
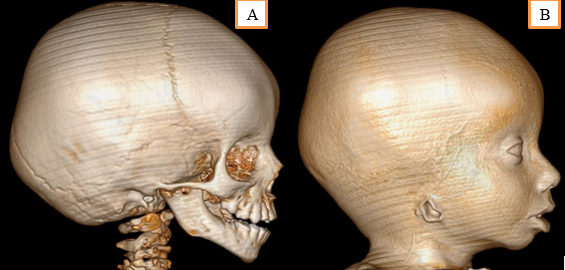
Micrognathia was present and the child had a deviation in mouth opening towards the affected side. On intraoral dental examination, there was cleft of palate involving the secondary palate (Figure 2). Patient had episodes of nasal regurgitation to solid foods. Dentition was examined and the child was diagnosed with Early Childhood Caries (ECC), followed by malocclusion due to hypoplastic mandible. On ophthalmic examination, in relation to right eye there was proptosis followed by inability to close the eyelid, resulting in conjuctivalxerosis, and trichosis. Corneal opacity was noted and it was sclerotic with superficial vascularization. Ocular dermoids over lateral limbus was noted in right eye. In left eye examination, the palpebral fissure was narrowed, pupillary reaction to light was present, cornea was clear and extraocular muscle movements were free and full. Fundus was dilated and evaluated under sedation, where the disc, vessels, media and macula of retina were normal in left eye. On aural examination, the child had no hearing defects. Child responded to sounds. There was aural atresia of right ear. The pinna was absent, while there was an opening of the external auditory canal which ends blindly covered by skin. Child was subjected to Brainstem Evoked Response Audiometry (BERA) and the impression was within normal limits. CT facial bones and neck was done to examine the mandible and cervical bones (Figure 3 A, B). Mandible was hypoplastic with well-defined condyle and coronoid processes. Maxilla had a bony defect in median region of hard palate associated with gross communication between the nasal and oral cavity (maximum transverse diameter 1.2cm) — cleft palate.
Discussion
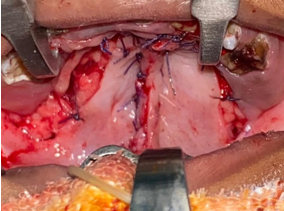
The Goldener syndrome is one amongst the rarest craniofacial anomalies with an incidence between the ratio of 1:3500 and 1:5600. The predilection of sex in this syndrome is inclined towards the male than female, with a ratio of 3:2.5 Most of the cases are reported to be sporadic, although it shows multifactorial heredity and also has presence of autosomal dominant genetic marker. Studies suggest that chromosome 22q11 consists of genes, which may alter the regulatory signaling events during the pharyngeal arch development. This error during the embryogenesis, contribute to the craniofacial dysmorphism.2 The etiopathogenesis of this syndrome is unclear and literature raises possibilities of abnormal pharyngeal arch (second arch in particular) formation during mesodermal migration and/or abnormal vascular supply during blastogenesis period (Poswill’s hypothesis). 1 Our case report supports the literature, with a unilateral presentation of the Goldenhar syndrome characteristics of ipsilateral facial, aural under development and ocular dermoid of right side face in a male patient. Prenatal diagnosis is possible, but however requires precise accuracy with ultrasound. In a sonographic study by De Catte et al., a 15-week foetus was observed with unilateral microphthalmia and maxillary cleft.6 Our patient was prenatally not diagnosed for any craniofacial abnormalities. Deoxyribonucleic acid testing cannot be performed as this syndrome is not gene specific. Treacher Collins Syndrome (TCS) is the closest related differential diagnosis for GS, but since TCS is gene specific (mutation of TCOF1 on chromosome 5q31-34) it helps in delineating the diagnosis. 7 Cases having familial history of GS due to consanguineous marriages have been reported, however our patient is the only child born from a non-consanguineous marriage. The clinical characteristics of GS consist of multiple malformations. Its clinical manifestations include ocular, aural, facial, vertebral and other systemic abnormalities.8 Apart from the classical features, Bowen et al. in 1971 described other variable abnormalities such as unilateral/bilateral macrosomia, malocclusion, bifid tongue, cleft palate, bregmatic patency, Teratology of Fallot, bundle branch block, epilepsy and recto-vaginal fistula.9 Our patient was presented with cleft palate and his cardiologic evaluation did not detect any cardiac abnormalities. Treatment for this syndromic condition is multidisciplinary and requires intervention at different stages of life depending on its severity and need for management. Aural defects can be addressed for cosmetic purposes if hearing is unaltered. Syndromes that have accessory tragi as a clinical feature are Townes–Brocks syndrome, Treacher–Collins syndrome, VACTERL syndrome and Wolf–Hirschhorn syndrome.10 Accessory tragi can be excised if needed. In ourcase, there was no hearing loss and hence we planned for a two staged helix reconstruction when the child turns 6-8 years. Ocular defects can be corrected for functional and aesthetic purposes. Cleft lip and palate was observed in 34% of the cases in a study by Hercíliomartelli-júnior et al., whereas usually it is seen only in 7-25% of the cases.11 Cleft palate repair was performed in this child under general anesthesia by Bardach’s two flap palatoplasty technique and the child had an uneventful post-operative period (Figure 4). At one month post-operative follow-up the complaints of nasal regurgitation have been completely resolved.
Conclusion
The purpose and critical analysis of this case report aids to understand and treat the craniofacial malformations and other general anomalies of Goldenhar syndrome in an early stage of the patient’s life. The advantages of early detection are not only beneficial for the treating surgeon/ physician, but also decrease the psychosocial difficulties for both the patient and their family. The importance to specify this rare syndrome is to achieve better prognostic outcomes in range of function and aesthetics. Multidisciplinary approach of treatment serves the best result and provides moral support to the patient’s kin and kith.